
Something tiny interrupted a lethal handshake inside brain cells. The consequence: mitochondria — the cell's power stations — began to fail, and neurons that once hummed with energy started to falter. That pairing of events is at the center of a new study from Case Western Reserve University School of Medicine, which traces a direct route from alpha-synuclein clumps to mitochondrial breakdown and offers a molecular bait to stop the damage.
Alpha-synuclein has been a suspect in Parkinson's disease for decades. When it misfolds and aggregates, neurons suffer. Separately, clinicians and lab scientists have observed that mitochondria in affected neurons are weak, energy-starved, and prone to triggering cell death. But how those two phenomena connect has been murky. The new work illuminates a precise biochemical interaction: alpha-synuclein hooks onto an enzyme deep inside mitochondria called ClpP, and that liaison appears to short-circuit the organelle's ability to manage damaged proteins and maintain metabolic balance.
What the team found and how they tested it
In laboratory tissue and animal models, the investigators watched alpha-synuclein bind to mitochondrial ClpP and noticed ensuing dysfunction in mitochondrial protein quality control. Think of ClpP as the garbage-disposal system in the powerhouse; when it's blocked, waste accumulates and the engine sputters. The downstream effects are familiar to anyone who studies Parkinson's: neurons lose their efficiency, dopamine-producing circuits decline, inflammation rises, and movement and cognition begin to suffer.
Rather than attacking alpha-synuclein wholesale, the researchers designed a short peptide named CS2 to act as a decoy. The idea is simple and elegant: lure the rogue protein away from ClpP so mitochondria can do their job. In ex vivo human brain tissue, in cultured neurons, and in mouse models, CS2 reduced markers of inflammation, preserved mitochondrial activity, and produced measurable improvements in motor and cognitive tests of the animals.
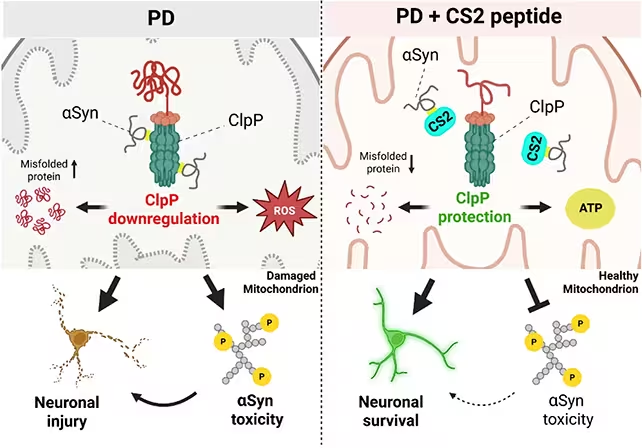
CS2 was shown to protect the ClpP enzyme.
That protective effect is significant because it targets a mechanistic step thought to be upstream of many of Parkinson's cellular failures. If alpha-synuclein’s interaction with ClpP is one of the early derailments in the disease cascade, then shielding ClpP could halt or slow multiple downstream harms.
Scientific context and implications
Parkinson's is not a single-pathway disorder. Genes, environment, aging, and cellular stressors mesh together in complex ways. This new study does not claim a universal cure. What it does offer is clarity: a defined molecular contact point that can be modified. Intervening here could be complementary to approaches that reduce alpha-synuclein aggregation, boost mitochondrial biogenesis, or modulate inflammation.
The research team estimates a five-year runway before CS2 or similar biologics could reach first-in-human trials. That's a reasonable timeline for a peptide-based therapy, but caution is warranted. Any molecule that alters protein interactions inside mitochondria must be rigorously screened for off-target effects. Small shifts in protein quality control can ripple into unexpected outcomes.
Still, the study shifts the tectonic plates of Parkinson's research just a little. It gives scientists a targetable mechanism and a tangible prototype molecule to refine. For families living with the disease, that incremental clarity is meaningful. Therapies that preserve neuronal energy metabolism could translate into longer, better-functioning lives.
Expert Insight
"This work is a rare combination of mechanistic precision and therapeutic imagination," says Dr. Elaine Ramirez, a fictional neurobiologist who studies mitochondrial dynamics. "Designing a decoy to intercept a pathogenic protein at the mitochondrial doorstep is clever. The challenge ahead is delivery — getting sufficient peptide into vulnerable neurons without weakening other cellular quality-control systems."
The paper appears in Molecular Neurodegeneration and opens several avenues: optimizing CS2's stability, testing long-term safety, and exploring whether similar decoys can protect other mitochondrial enzymes. It also prompts a broader question: if we can prevent mitochondria from losing power, might we change Parkinson's trajectory from a relentlessly progressive decline to a condition that can be managed or even arrested? The experiments ahead will be decisive, but for now the field has a fresh lead to follow.

“The cosmos has always fascinated me. I write about space missions, astronomy, and the technologies pushing humanity beyond Earth.”

Discussion
Leave a Comment